2026-05-19
Nuclear Drug: 225Ac-ch806
Journal of Nuclear Medicine 2024; 00:1–7
This article analyzes the key data of preclinical studies on 225Ac nuclear drugs with examples. Published in December 2024 in the Journal of Nuclear Medicine (JNM), this study investigates a novel tumor-specific antibody for Epidermal Growth Factor Receptor (EGFR), ch806, labeled with 225Ac. It evaluates its therapeutic efficacy in Glioblastoma and Colorectal Cancer xenograft models. This research paper is rich in content and can serve as a model to deeply analyze the core points and specific details of such studies.
Step 1: Selecting the Target
Epidermal Growth Factor Receptor (EGFR) is a transmembrane receptor tyrosine kinase and a member of the HER (human epidermal growth factor receptor) family. EGFR is overexpressed or mutated in various epithelial tumors, such as Non-Small Cell Lung Cancer (NSCLC), Head and Neck Squamous Cell Carcinoma, and Colorectal Cancer. EGFR activates its tyrosine kinase activity by binding with ligands, thereby activating downstream signaling pathways such as RAS-RAF-MEK (MAPK/ERK), PI3K-AKT-mTOR, and JAK/STAT, regulating cell proliferation, differentiation, migration, and survival. The presence of EGFR mutations makes tumors more sensitive to specific targeted therapeutic drugs, such as the first-generation EGFR Tyrosine Kinase Inhibitors (TKIs) Erlotinib and Gefitinib. However, almost all patients receiving EGFR-TKIs will eventually develop resistance, with the most common mechanism being the T790M mutation. Because of this, research on EGFR-targeted therapeutic drugs has progressed rapidly, reaching the fourth generation.
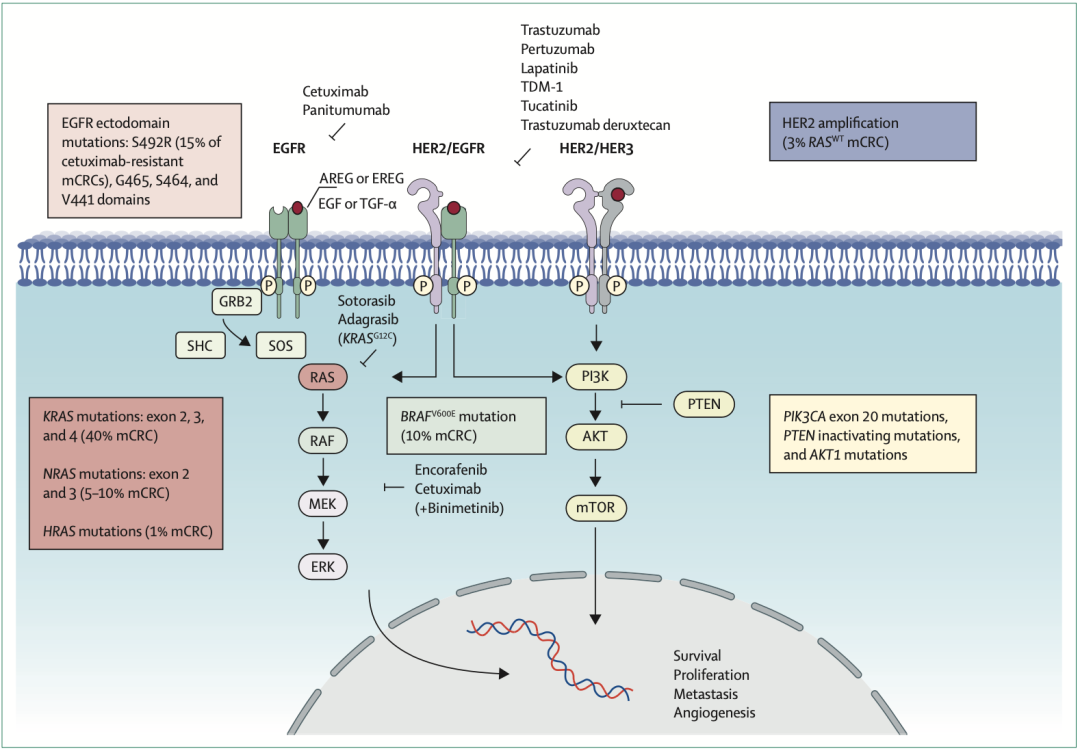 Example Figure: This figure illustrates the Epidermal Growth Factor Receptor (EGFR) signaling pathway and its role in colorectal cancer. The EGFR signaling pathway plays a critical role in the development and progression of colorectal cancer, involving multiple molecules and steps.
Example Figure: This figure illustrates the Epidermal Growth Factor Receptor (EGFR) signaling pathway and its role in colorectal cancer. The EGFR signaling pathway plays a critical role in the development and progression of colorectal cancer, involving multiple molecules and steps.
Paper Title: Targeting the EGFR signalling pathway in metastatic colorectal cancer.
Existing EGFR-TKIs have achieved certain efficacy in treating EGFR-mutant tumors, but the development of resistance limits their long-term application. Furthermore, for tumors that have already developed resistance, especially those with complex mutation profiles such as T790M and C797S mutations, the effects of existing treatments are limited. Therefore, developing novel therapeutic strategies such as nuclear drugs may provide new treatment options for these patients. Radioimmunotherapy, as an emerging treatment modality, has potential synergistic effects and may be combined with existing targeted and immunotherapies to provide a more comprehensive treatment plan.
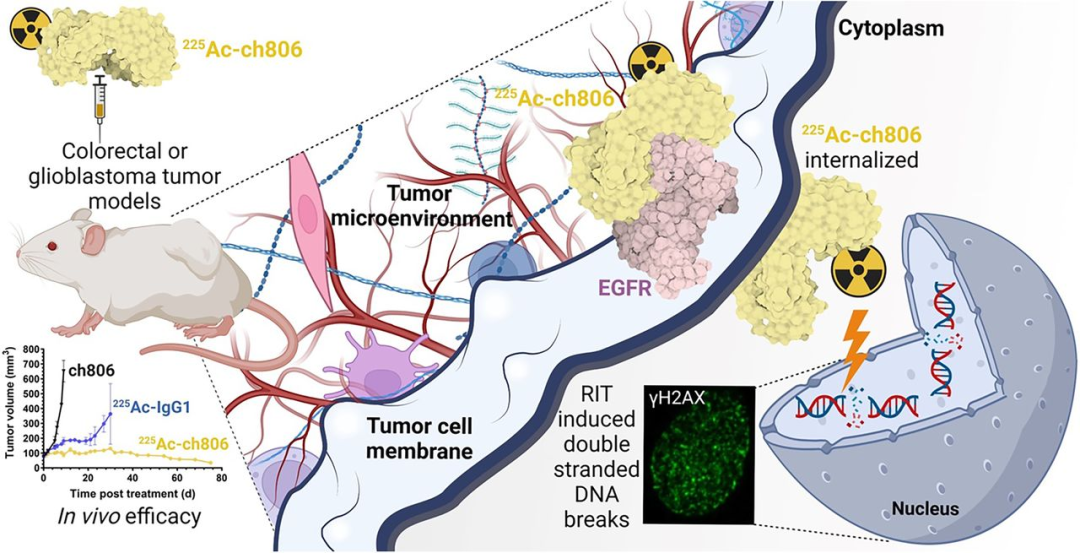
Paper Title: Radiolabeling and Preclinical Evaluation of Therapeutic Efficacy of 225Ac-ch806 in Glioblastoma and Colorectal Cancer Xenograft Models.
In the study, 225Ac-ch806 was prepared using different chelators, including [225Ac]Ac-macropa-tzPEG3Sq-ch806 and [225Ac]Ac-DOTA-dhPzPEG4-ch806. The research team quantified the radiochemical yield, purity, apparent specific activity, and serum stability of 225Ac-ch806. They systematically evaluated the in vitro cell killing effect and studied the biodistribution and therapeutic effect of 225Ac-ch806 in mice bearing U87MG.de2-7 and DiFi tumors. Pharmacodynamic analysis was performed on tumors after treatment, including gH2AX detection for DNA double-strand breaks via immunofluorescence, and immunohistochemical analysis of proliferation, cell cycle arrest, and apoptosis.
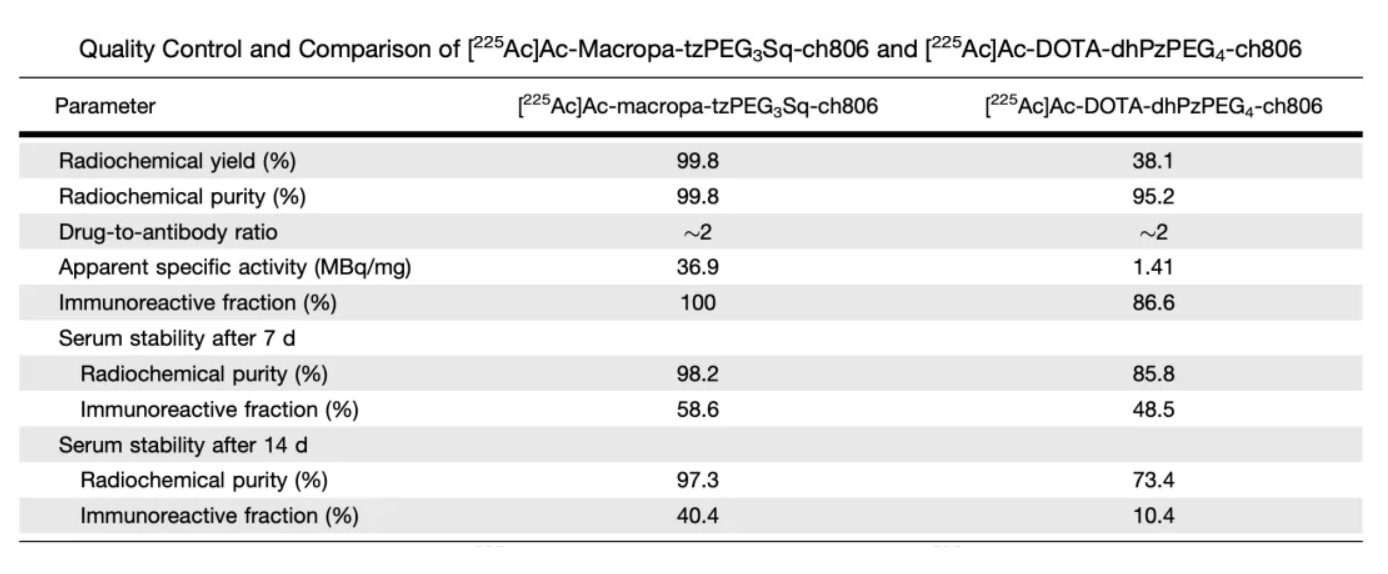 Table 1 presents comparative data for 225 Ac-ch806 labeled with two different chelators, including radiochemical yield, radiochemical purity, drug-to-antibody ratio (DAR), apparent specific activity, immunoreactive fraction, and serum stability.
Table 1 presents comparative data for 225 Ac-ch806 labeled with two different chelators, including radiochemical yield, radiochemical purity, drug-to-antibody ratio (DAR), apparent specific activity, immunoreactive fraction, and serum stability.
First is the preparation and characterization assessment of 225Ac-ch806:
By combining the ch806 antibody with different chelators, two different 225Ac-ch806 compounds were prepared: [225Ac]Ac-macropa-tzPEG3Sq-ch806 and [225Ac]Ac-DOTA-dhPzPEG4-ch806. Subsequently, the radiochemical yield, purity, apparent specific activity, and serum stability were evaluated. The results showed that [225Ac]Ac-macropa-tzPEG3Sq-ch806 is superior to [225Ac]Ac-DOTA-dhPzPEG4-ch806 in terms of radiochemical yield, purity, apparent specific activity, and serum stability. Therefore, [225Ac]Ac-macropa-tzPEG3Sq-ch806 (i.e., 225Ac-ch806) was selected for subsequent in vitro and in vivo studies.
Next is the in vitro cell killing effect test:
The in vitro cell killing effect of 225Ac-ch806 was assessed by co-culturing U87MG.de2-7 cells with ch806, free 225Ac, 225Ac-ch806, and 225Ac-IgG1 for 4 days. Compared to ch806 and free 225Ac, 225Ac-ch806 showed significant, specific, and dose-dependent anti-proliferative effects, reducing cell viability.
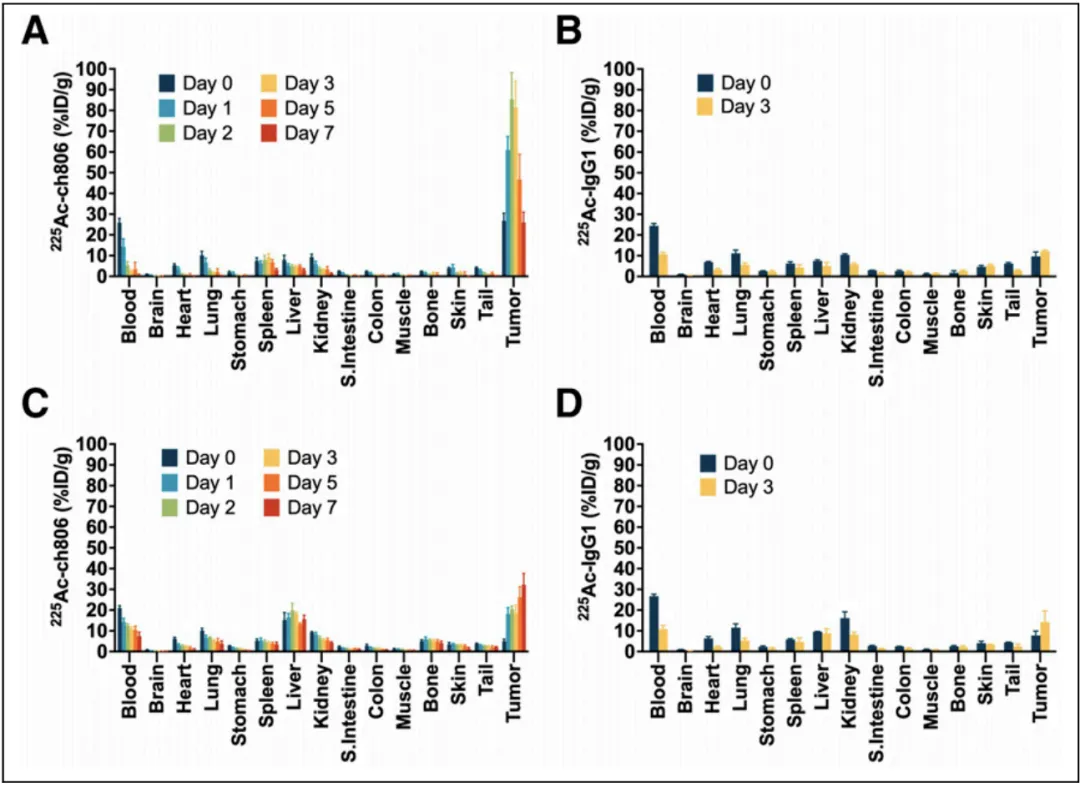
This figure illustrates the biodistribution of 225Ac-ch806 and 225 Ac-IgG1 in U87MG.de2-7 (glioblastoma) and DiFi (colorectal cancer) xenograft models. The data show that tumor uptake of 225 Ac-ch806 peaked on day 2 post-injection in the U87MG.de2-7 model, whereas tumor accumulation gradually increased until day 7 in the DiFi model. These results highlight the tumor-specific targeting capability of 225 Ac-ch806, as well as its potential therapeutic application across different tumor models.
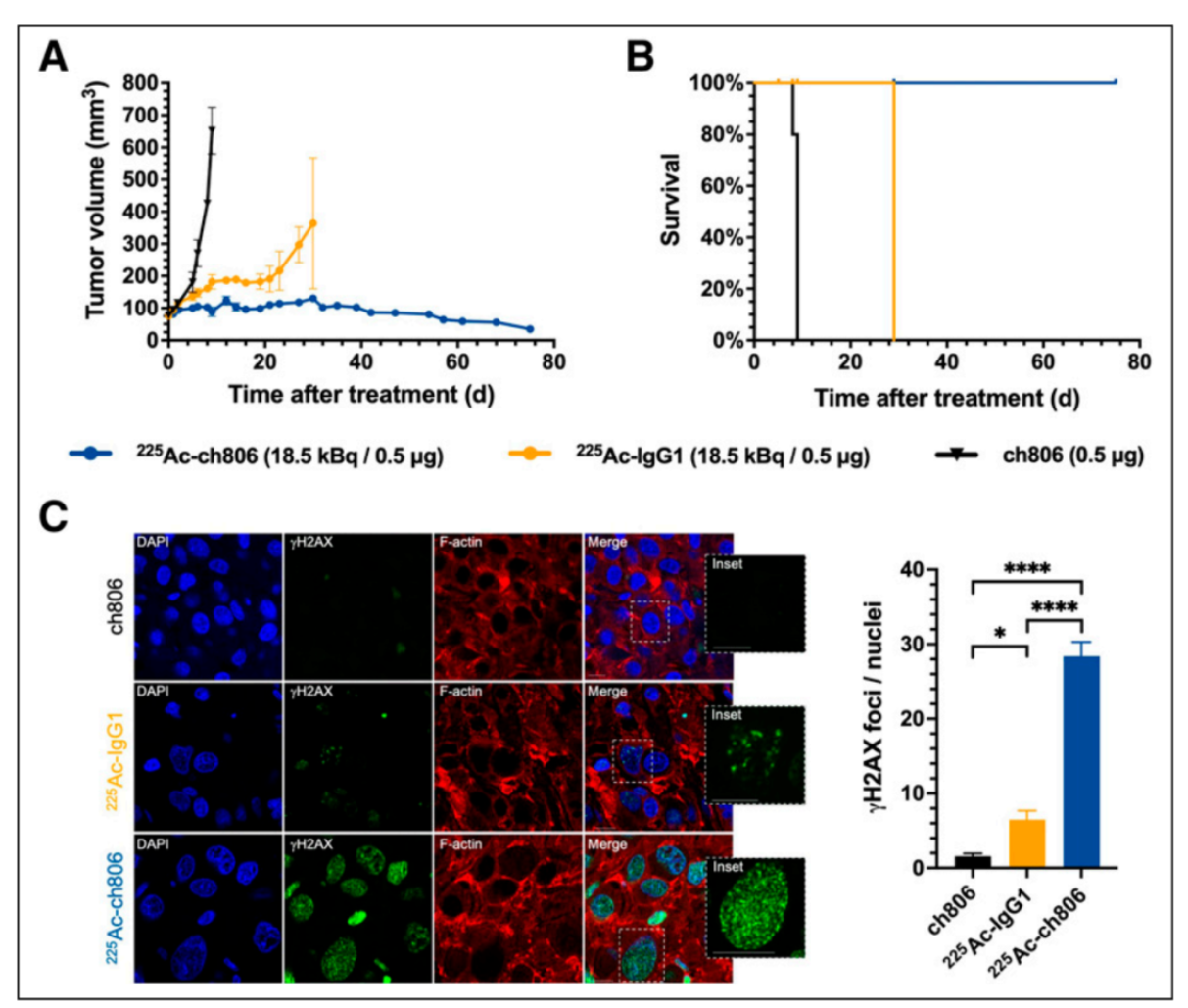
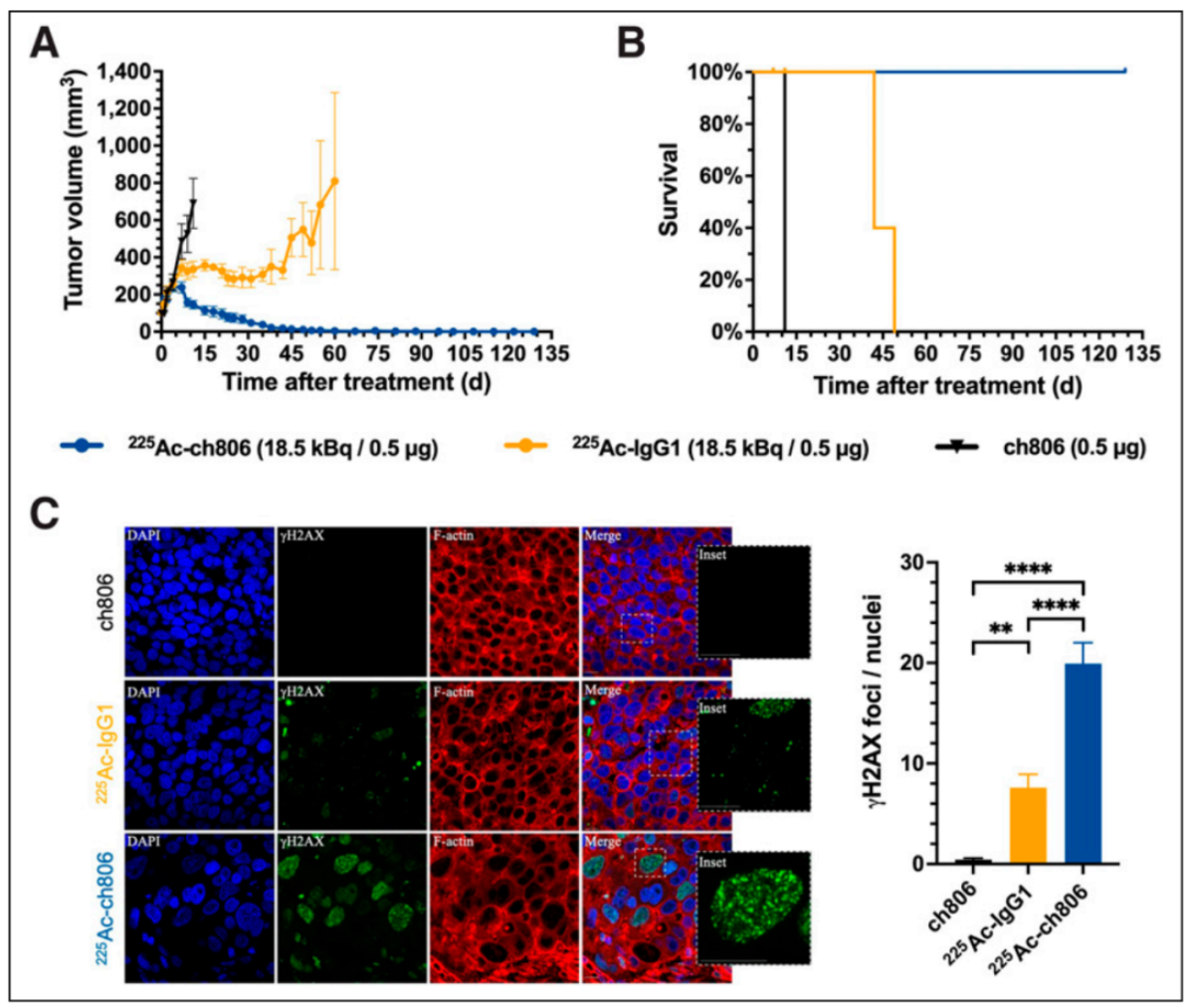 This figure presents the therapeutic outcomes of 225 Ac-ch806 in the DiFi model, including tumor growth curves, survival curves, and immunofluorescence staining for γγ H2AX. Despite relatively low accumulation of 225 Ac-ch806 in DiFi tumors, mice treated with 225 Ac-ch806 showed a steady decline in tumor volume, with 7 mice achieving a complete response and 1 showing a partial response. The survival curve demonstrated prolonged survival in the 225 Ac-ch806-treated group. Additionally, higher levels of DNA double-strand breaks were observed in treated tumors compared to the control group, further confirming the therapeutic efficacy of 225 Ac-ch806.
This figure presents the therapeutic outcomes of 225 Ac-ch806 in the DiFi model, including tumor growth curves, survival curves, and immunofluorescence staining for γγ H2AX. Despite relatively low accumulation of 225 Ac-ch806 in DiFi tumors, mice treated with 225 Ac-ch806 showed a steady decline in tumor volume, with 7 mice achieving a complete response and 1 showing a partial response. The survival curve demonstrated prolonged survival in the 225 Ac-ch806-treated group. Additionally, higher levels of DNA double-strand breaks were observed in treated tumors compared to the control group, further confirming the therapeutic efficacy of 225 Ac-ch806.
 This figure presents immunohistochemical analyses of U87MG.de2-7 and DiFi tumors using Hematoxylin and Eosin (H&E) staining, as well as Ki-67, p21, and cleaved caspase 3 staining, to assess tumor architecture, proliferation capacity, cell cycle, and apoptosis. Compared with the control group, 225 Ac-ch806-treated tumors exhibited reduced Ki-67 staining, indicating decreased proliferation. In the U87MG.de2-7 model, p21 expression was significantly upregulated by 225 Ac-ch806, suggesting potential cell cycle arrest induced by DNA double-strand breaks. In the DiFi model, 225 Ac-ch806-treated tumors showed significantly increased levels of apoptosis, indicating that 225 Ac-ch806 promotes cell death via the apoptotic pathway.
This figure presents immunohistochemical analyses of U87MG.de2-7 and DiFi tumors using Hematoxylin and Eosin (H&E) staining, as well as Ki-67, p21, and cleaved caspase 3 staining, to assess tumor architecture, proliferation capacity, cell cycle, and apoptosis. Compared with the control group, 225 Ac-ch806-treated tumors exhibited reduced Ki-67 staining, indicating decreased proliferation. In the U87MG.de2-7 model, p21 expression was significantly upregulated by 225 Ac-ch806, suggesting potential cell cycle arrest induced by DNA double-strand breaks. In the DiFi model, 225 Ac-ch806-treated tumors showed significantly increased levels of apoptosis, indicating that 225 Ac-ch806 promotes cell death via the apoptotic pathway.
Refining the Pharmacodynamic Analysis:
By implementing Hematoxylin and Eosin staining and immunohistochemical analysis of Ki-67, p21, and cleaved caspase 3, researchers assessed the structure, proliferation, cell cycle, and apoptosis of U87MG.de2-7 and DiFi tumors. Compared to the control group, tumors treated with 225Ac-ch806 showed increased DNA double-strand breaks, decreased Ki-67 expression (indicating reduced proliferation), and significant upregulation of p21 expression in the U87MG.de2-7 model, which may lead to cell cycle arrest by inducing DNA double-strand breaks. For the DiFi model, tumors treated with 225Ac-ch806 showed a significant increase in apoptosis levels, suggesting that 225Ac-ch806 can induce cell death by promoting apoptotic pathways.
Step 3: Organizing Experimental Data to Draw Conclusions
After a series of detailed experiments and analysis, the research results clearly point out that in Glioblastoma and Colorectal Tumor models, 225Ac-ch806 can effectively inhibit tumor growth by causing DNA double-strand breaks, thereby restricting cancer cell proliferation, and triggering cell cycle arrest and cell apoptosis. These important findings highlight the great potential of 225Ac-ch806 as a potential clinical therapeutic strategy for EGFR-expressing solid tumors.
Conclusion
Although there are relatively few EGFR-targeted nuclear drugs in research and development, recent research progress indicates that this field is expected to achieve major breakthroughs. 64Cu-NCAB001, developed by the National Institutes for Quantum Science and Technology (QST) in Japan, is undergoing a Phase I clinical study. Its preclinical study results provide strong support for the PET clinical trial of this drug, suggesting it may become a powerful tool for the early diagnosis of pancreatic cancer. Furthermore, Peking University Cancer Hospital initiated a clinical IIT study of 18F-LF13 (NCT06641674) in October 2024, further promoting the research on EGFR-targeted nuclear drugs. The anti-EGFR/c-Met antibody-conjugated radionuclide drug [225Ac]225Ac-FPI-2068, co-developed by Fusion Pharmaceuticals and AstraZeneca, had its Phase 1 clinical study (NCT06147037) publicized in November 2023. This is a non-randomized, multicenter, open-label study designed to evaluate the safety, tolerability, dosimetry, biodistribution, and pharmacokinetics of [225Ac]225Ac-FPI-2068, [111In]In-FPI-2107, and FPI-2053 in patients with metastatic and/or recurrent solid tumors. These studies cover various tumor types such as Head and Neck Squamous Cell Carcinoma, Non-Small Cell Lung Cancer, Metastatic Colorectal Cancer, and Pancreatic Ductal Adenocarcinoma, providing new perspectives and hope for the research and application of EGFR-targeted nuclear drugs. We look forward to EGFR-targeted new nuclear drugs serving patients faster and better, bringing significant improvements to their health and quality of life.
Address:Ping An Fortune Center, Lize Financial Business District of Beijing







