2026-05-20
As a hot target in tumor immunotherapy, B7-H3 has not only attracted fierce competition among multiple ADC (Antibody-Drug Conjugate) developers but has also garnered significant attention due to the tumultuous commercialization journey of one specific radiopharmaceutical—Omburtamab. From the technology transfer at MSK and the FDA's unanimous rejection based on a single-arm trial, to pipeline restructuring and continued exploration, this case reflects the complex interplay between scientific rigor and commercial strategy in new drug development. Notably, such narratives are far from isolated incidents in the global innovative drug landscape...
B7-H3 (CD276) is a key member of the B7 immune checkpoint family. As a transmembrane immunomodulatory molecule, it exhibits distinct pathological specificity within the tumor microenvironment. Studies indicate that B7-H3 is specifically expressed in various malignant tumor cells and tumor-infiltrating immune cells (e.g., monocytes, dendritic cells, myeloid-derived suppressor cells [MDSCs], neutrophils, macrophages, B cells, and activated T cells), while its expression in normal tissues is extremely low and strictly restricted. This significantly dysregulated expression pattern is closely associated with tumor immune evasion, malignant proliferation, invasion, and metastasis. Consequently, B7-H3 serves not only as a critical biomarker for assessing tumor malignancy but also as a highly promising therapeutic target due to its high selectivity in tumor tissues.
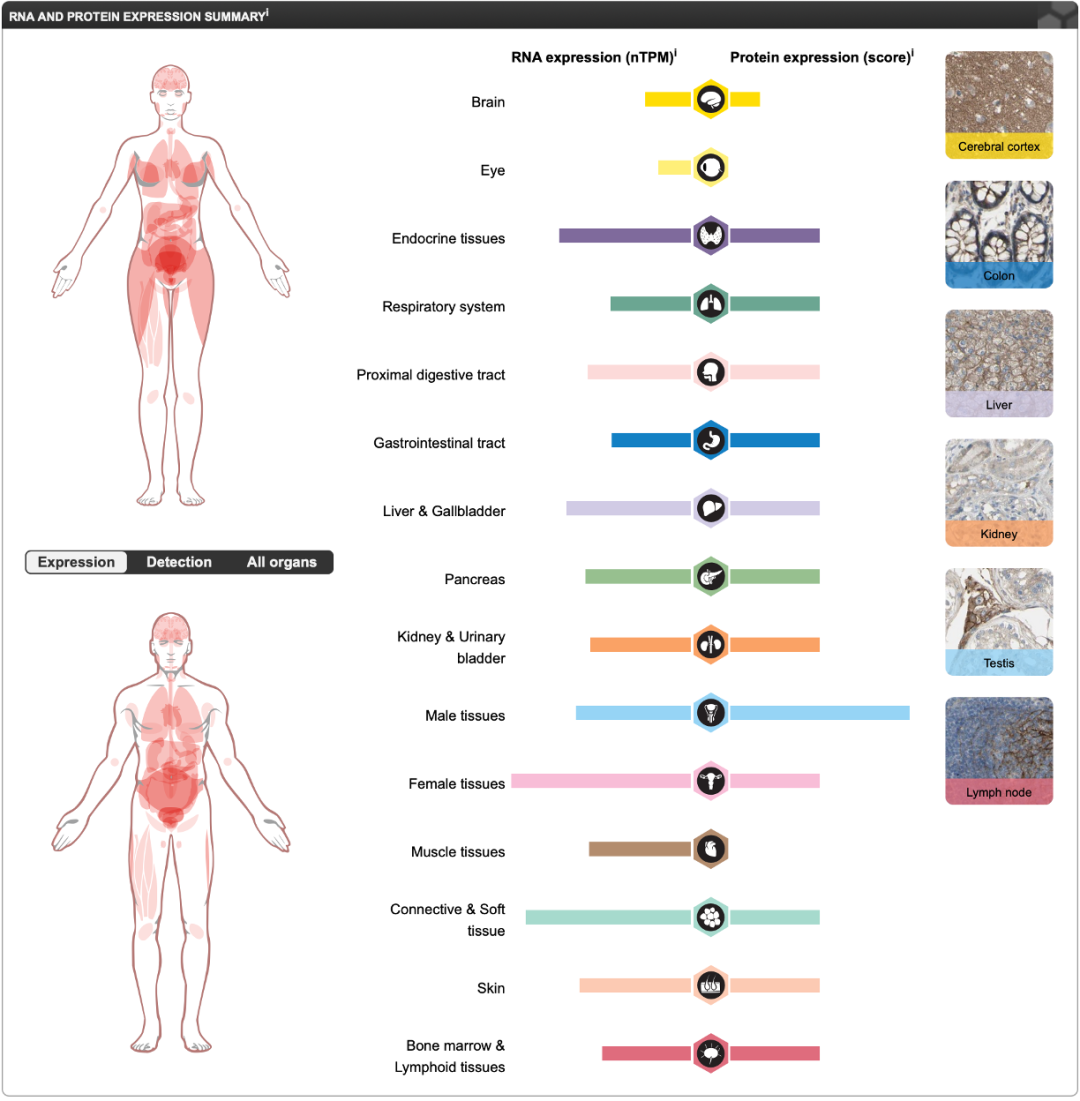
Expression Distribution of B7-H3:
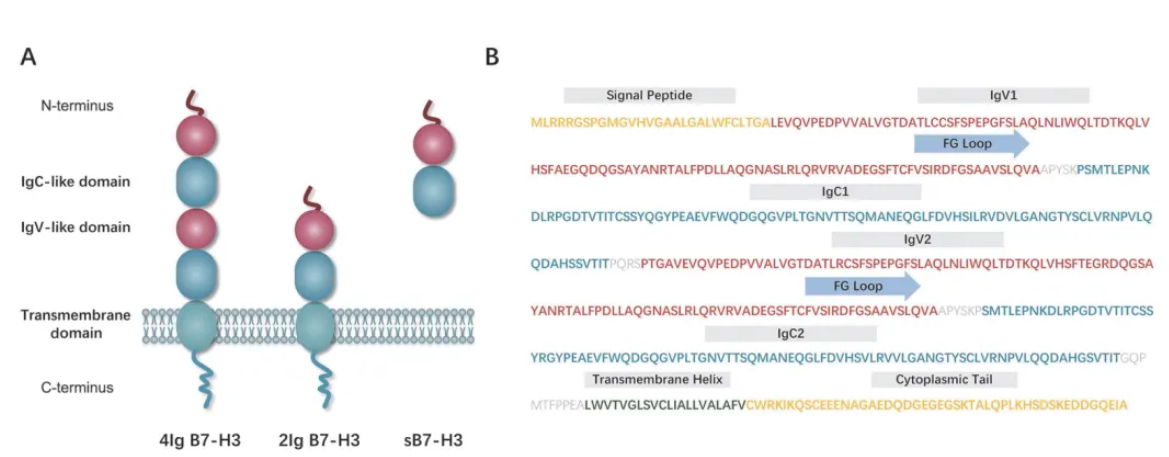
Structure of B7-H3:
B7-H3 (CD276) is a type I transmembrane glycoprotein belonging to the B7 immune checkpoint family. Its structure consists of three parts: an extracellular domain, a transmembrane domain, and a highly diverse intracellular domain.
 Data Source: https://www.proteinatlas.org
Data Source: https://www.proteinatlas.org
Functions and Disease Associations of B7-H3:
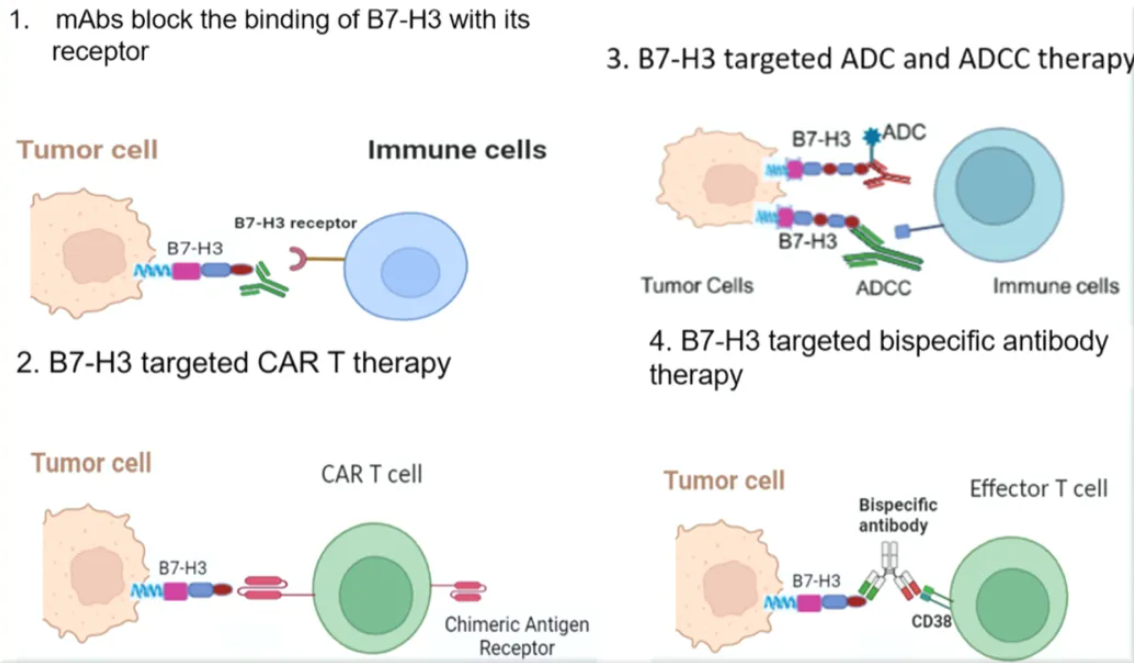
Its oncogenic mechanism presents dual characteristics. On one hand, B7-H3 constructs an immunosuppressive microenvironment by inhibiting the cytotoxic activity of T cells, hindering interferon-gamma (IFN-γ) secretion, promoting regulatory T cell (Treg) differentiation, and weakening natural killer (NK) cell function. On the other hand, its non-immune-dependent functions can activate oncogenic signaling pathways such as JAK/STAT and PI3K/AKT, driving tumor cell proliferation, epithelial-mesenchymal transition (EMT), and angiogenesis, while simultaneously enhancing tumor resistance to chemotherapy and distant metastatic capability. Notably, monoclonal antibodies, bispecific antibodies, and antibody-drug conjugates (ADCs) developed against specific glycosylation epitopes of B7-H3 have demonstrated significant anti-tumor activity in preclinical studies. Furthermore, CAR-T therapies targeting B7-H3 have achieved breakthrough progress in the treatment of solid tumors. These advancements position B7-H3 as one of the most translationally valuable molecular targets in current tumor immunotherapy.
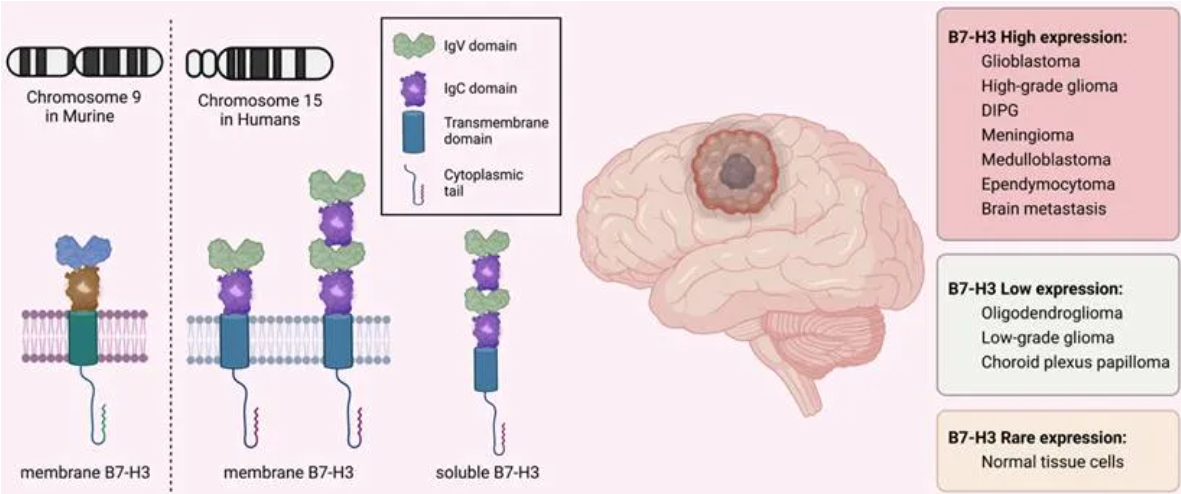 Data Source: Int J Biol Sci 2023; 19(12):3762-3780
Data Source: Int J Biol Sci 2023; 19(12):3762-3780
As the world's first B7-H3-targeting Radiopharmaceutical Drug (RDC), the development journey of Omburtamab exemplifies a paradigm of strategic collaboration in innovative drug development. Developed by Memorial Sloan Kettering Cancer Center (MSK) based on its breakthrough antibody 8H9, the drug achieves precise radiotherapy for Central Nervous System/Leptomeningeal Metastatic Neuroblastoma (CNS/LM) through Iodine-131 labeling. In 2015, Y-mAbs Therapeutics reached a global exclusive licensing agreement with MSK, officially initiating the clinical translation of this project.
Based on its potential to address unmet needs in pediatric oncology, the drug received early global regulatory endorsement: In March 2017, the EMA granted it Orphan Drug Designation, providing 10 years of market exclusivity for the leptomeningeal metastasis neuroblastoma indication. In June of the same year, the FDA further granted Breakthrough Therapy Designation, advancing its application in second-line and subsequent treatments for pediatric neuroblastoma via an accelerated review pathway. These milestones, coupled with the exclusive licensing cooperation in Greater China signed with Sinocelltech in 2020, not only made Omburtamab a benchmark case for multinational pharmaceutical companies positioning in Asian radiopharmaceuticals but also thrust the B7-H3 targeted radiotherapy track into the industry spotlight. At that time, with dazzling clinical data and rising capital enthusiasm, it appeared poised to be a game-changer in the field of pediatric targeted tumor therapy.
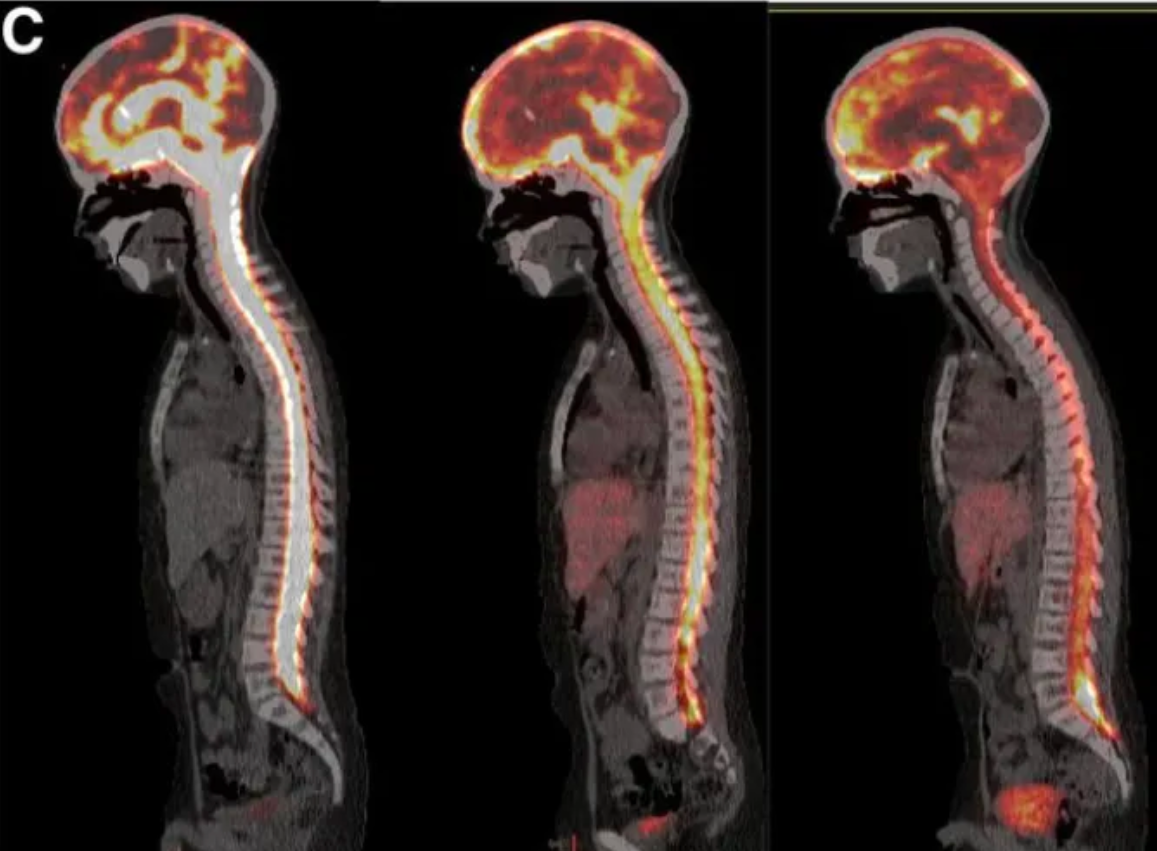 Data Source: Journal of Nuclear Medicine, December 2019, 60(12): 1794-1801; Serial ¹²⁴I-omburtamab images in a patient with metastatic neuroblastoma and leptomeningeal disease: Fused images from D0, D1, and D2 show decreasing activity over time within the ventricles and cerebrospinal fluid pathways.
Data Source: Journal of Nuclear Medicine, December 2019, 60(12): 1794-1801; Serial ¹²⁴I-omburtamab images in a patient with metastatic neuroblastoma and leptomeningeal disease: Fused images from D0, D1, and D2 show decreasing activity over time within the ventricles and cerebrospinal fluid pathways.
However, its development journey became a classic cautionary tale regarding clinical design in innovative drugs. In October 2022, the FDA raised scientific concerns regarding the selection of historical control groups in the single-arm trial—patient data spanning a decade contained key variable biases such as geography (USA, Germany) and lines of therapy. As an external clinical control for a single-arm study, the data could not provide a reasonable comparison across time, location, and prior treatment lines. Following survival benefit analysis, the FDA Advisory Committee deemed the efficacy evidence insufficient, ultimately closing the approval pathway with a unanimous 16:0 vote against approval and a subsequent Complete Response Letter (CRL). This setback not only led to the termination of Omburtamab's multi-nuclide exploration path (including the ¹²⁴I PET diagnostic product and ¹⁷⁷Lu therapeutic product) but also triggered severe volatility in the capital market, causing a cliff-like plunge in stock price and evaporating over 80% of its market value...
On January 18, 2023, investors filed a class-action lawsuit demanding compensation for investment losses caused by misleading conduct. Investors alleged that the company exaggerated the likelihood of FDA approval for the drug and failed to adequately disclose the risk that the drug might not be approved. Such misleading behavior potentially led investors to make decisions based on incorrect information. Ultimately, Y-mAbs agreed to pay $19.65 million to settle with shareholders, resolving the litigation. This indicates the company acknowledged potential misconduct in information disclosure and was willing to offer financial compensation to appease investor dissatisfaction.
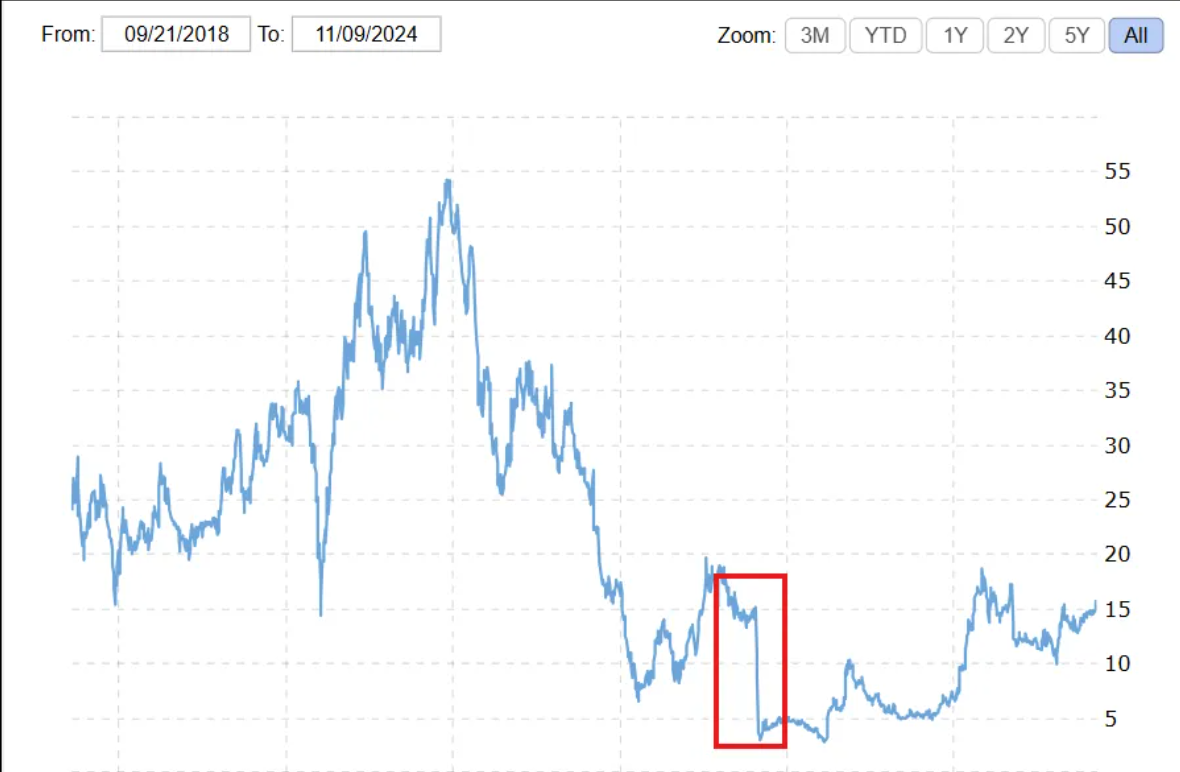 Y-mAbs Stock Decline - October 26, 2022
Y-mAbs Stock Decline - October 26, 2022
A dramatic turning point emerged following strategic restructuring: The company decisively divested non-core assets (including the GD2-GD3 vaccine and CD33 bispecific antibody) and streamlined its team by 35%, extending its cash runway to 2027. Simultaneously, R&D focus shifted toward the nuclear drug delivery technology SADA (Stepwise Avidin-Dotin System, a pretargeted radioimmunotherapy technology; see image below for principle and current pipeline composition). This platform utilizes modular design to separate "pretargeting" from "nuclide therapy," significantly reducing radiation damage to normal tissues. Its first GD2-SADA project initiated Phase 1 clinical trials for solid tumors in 2023. With the radiopharmaceutical sector becoming a hotspot for MNC M&A (e.g., BMS acquiring RayzeBio for $4.1 billion), combined with cash flow support from Danyelza (GD2 mAb) sales growing 71% YoY to $84.3 million, and Y-mAbs announcing the appointment of Dr. Mary Tagliaferri (formerly of RayzeBio) to the board in 2024, the capital market has reignited expectations for Y-mAbs' technology translation, driving a rebound in its stock price.
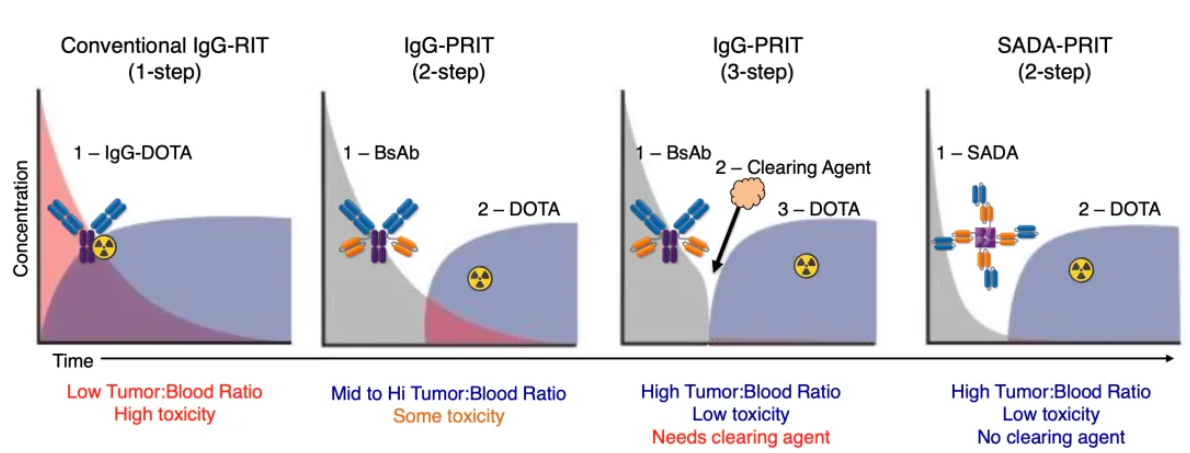 Data Source: Y-mAbs Therapeutics Official Website
Data Source: Y-mAbs Therapeutics Official Website
The application for ¹³¹I-omburtamab relied primarily on two single-arm studies: Study 03-133 and Study 101. Detailed introductions follow:
Study 03-133(NCT00089245)
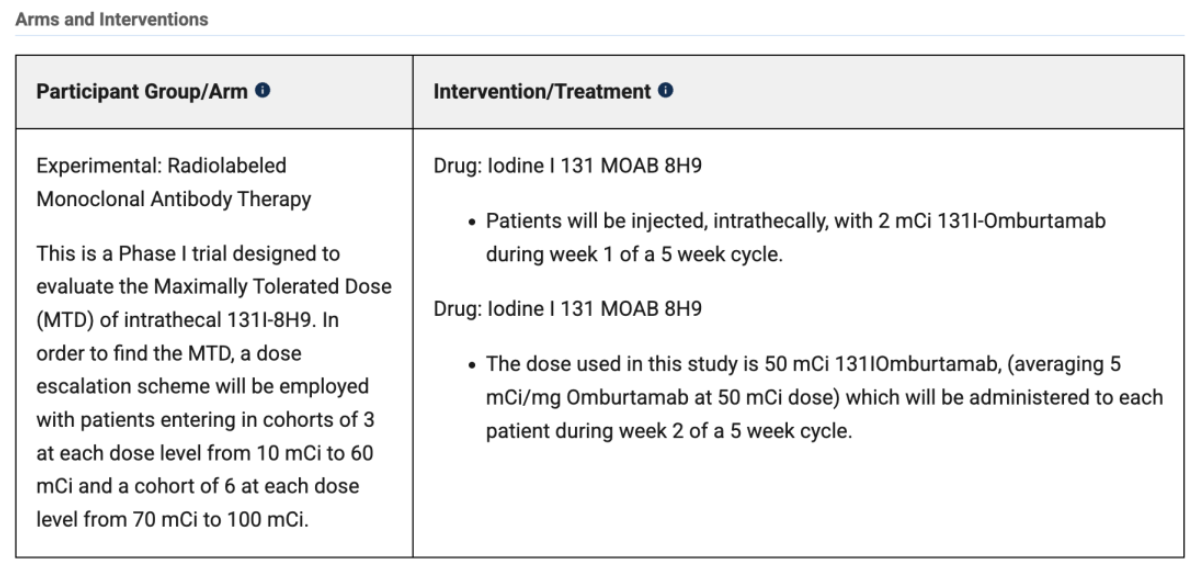
Design: A single-arm, single-center Phase I trial designed to explore the safe dose and preliminary efficacy of ¹³¹I-omburtamab in patients with relapsed/refractory neuroblastoma with central nervous system (CNS)/leptomeningeal metastases.

Kim Kramer's team published a Phase I clinical study in 2022, which utilized a 3+3 dose-escalation design to evaluate the safety of intraventricular administration of ¹³¹I‑omburtamab in 38 patients with B7H3-positive central nervous system (CNS) tumors. The study cohort included metastatic neuroblastoma (16 cases) and other B7H3-positive solid tumors (22 cases), with 35 patients completing at least one treatment cycle (including dosimetry and therapeutic administration).
Safety analysis showed that treatment-related acute toxicities were primarily self-limiting Grade 1-2 headaches, vomiting, and fever, accompanied by some biochemical abnormalities; Grade 3-4 thrombocytopenia was the most common hematologic toxicity. The recommended Phase II dose (RP2D) was determined to be 1850 MBq per administration. Pharmacokinetic data revealed a significant difference in radiation doses between the cerebrospinal fluid (1.01 mGy/MBq) and blood (0.04 mGy/MBq), highlighting the targeted therapeutic advantage. Radiation exposure to key organs remained below safety thresholds. Notably, patients who tested positive for anti-drug antibodies exhibited accelerated serum clearance, resulting in a significantly expanded therapeutic window compared to those without antibody production.
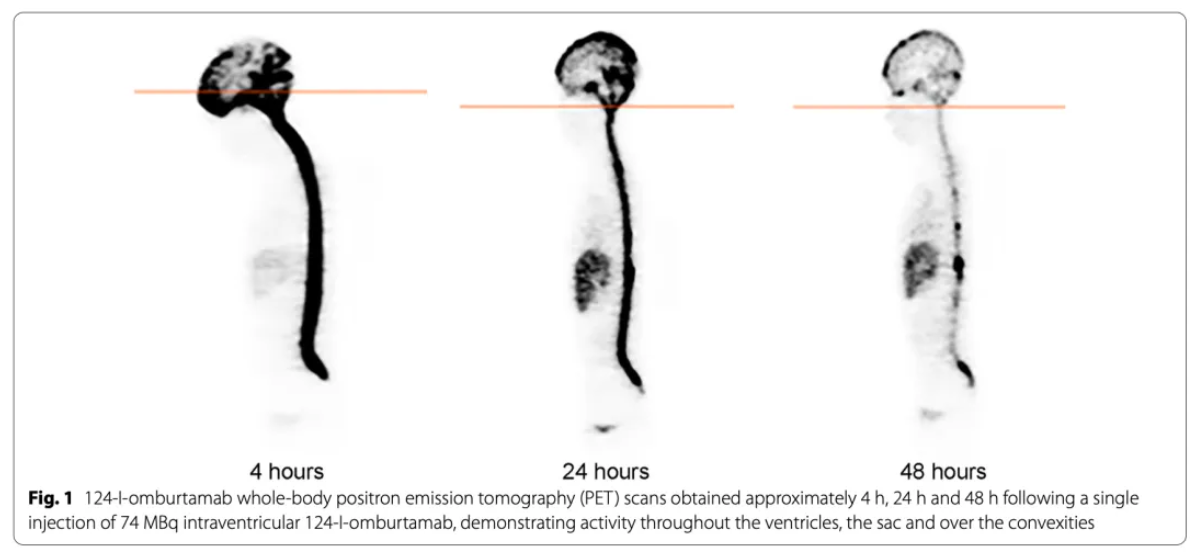 Data Source: https://doi.org/10.1186/s13045-022-01383-4; This image illustrates the dynamic distribution of the radiopharmaceutical 124I-omburtamab following intraventricular injection, revealing the diffusion patterns of the drug within the cerebrospinal fluid (CSF) system and its time-dependent clearance characteristics.
Data Source: https://doi.org/10.1186/s13045-022-01383-4; This image illustrates the dynamic distribution of the radiopharmaceutical 124I-omburtamab following intraventricular injection, revealing the diffusion patterns of the drug within the cerebrospinal fluid (CSF) system and its time-dependent clearance characteristics.
Through survival data comparison, long-term follow-up results, and statistical analysis, the study strongly supported that CRIT combined with radiotherapy could significantly improve the prognosis of patients with CNS-relapsed neuroblastoma, breaking through the survival bottleneck of traditional treatments:
Despite these findings, the FDA's stance on its marketing application was critical. Due to the use of an external control, the FDA considered there to be significant differences between the study population and the control group in terms of treatment intensity (e.g., radiotherapy, chemotherapy regimens), geography (USA vs. Germany), and time (long study span). Consequently, the survival difference could not be reliably attributed to the drug.
Study 101(NCT03275402)
 In October 2020 (interim analysis), Y-mAbs announced key clinical data updates at the International Society of Paediatric Oncology (SIOP) annual meeting: a multicenter study based on 17 patients showed a 12-month OS rate of 87% (median follow-up of 26 weeks), representing a breakthrough improvement over the historical control group (approx. 30%). In September 2022 (updated data), Dr. Kim Kramer from MSK presented results at the SIOP annual meeting: follow-up data expanded to 32 patients showed a 12-month OS of 73.5% (median follow-up of 25 months), confirming long-term survival benefits. Regarding safety, 40.6% of patients experienced serious adverse events (SAEs), primarily bone marrow suppression, but no uncontrollable toxicities or treatment-related deaths were reported.
In October 2020 (interim analysis), Y-mAbs announced key clinical data updates at the International Society of Paediatric Oncology (SIOP) annual meeting: a multicenter study based on 17 patients showed a 12-month OS rate of 87% (median follow-up of 26 weeks), representing a breakthrough improvement over the historical control group (approx. 30%). In September 2022 (updated data), Dr. Kim Kramer from MSK presented results at the SIOP annual meeting: follow-up data expanded to 32 patients showed a 12-month OS of 73.5% (median follow-up of 25 months), confirming long-term survival benefits. Regarding safety, 40.6% of patients experienced serious adverse events (SAEs), primarily bone marrow suppression, but no uncontrollable toxicities or treatment-related deaths were reported.
In December 2022, the FDA issued a Complete Response Letter (CRL) pointing out core deficiencies in Y-mAbs' marketing application for ¹³¹I‑omburtamab:

The FDA emphasized that existing data were difficult to support regulatory approval, requiring supplementary multicenter randomized controlled trials to confirm efficacy, optimized pharmacokinetic studies, and long-term follow-up to verify safety—especially the need to establish a stricter benefit-risk framework in pediatric patients.
Fundamentally, this outcome may have been inevitable. Since 2016, the FDA had repeatedly warned Y-mAbs that the patient population submitted in its clinical trials lacked comparability with the Childhood Brain Tumor Consortium Registry (CGCCR) population, and this was not a "new finding" as the company claimed. In the January 2022 discussion on resubmitting the BLA, the FDA reiterated that the CGCCR data had fundamental flaws (e.g., lack of a control group for patients receiving craniospinal irradiation), and Y-mAbs' analysis was criticized by the FDA as "arbitrary" and failing to adequately address key concerns. Despite the FDA explicitly requesting a reliable control or the initiation of an "alternative clinical development plan," Y-mAbs continued to claim to investors that it was "aligned with the FDA," possessed "complete data," and that the "application was progressing smoothly," attempting to mask substantial disagreements in regulatory communications. Y-mAbs positioned omburtamab as a disruptive therapy for CNS tumors, gambling between commercial ambition and scientific rigor. Over-reliance on single-arm data became a strategic misjudgment, reflecting the eternal contradiction in new drug development among speed, cost, and strength of evidence.
Although the drug has not yet been approved due to flaws in the single-arm trial design, it still retains possibilities for the clinical translation of B7-H3 targeted therapies. To this day, Y-mAbs continues to explore the clinical value of omburtamab, such as expanding indications and conducting precise stratification. Currently, the research scope has been extended from leptomeningeal metastasis of neuroblastoma to Diffuse Intrinsic Pontine Glioma (DIPG). For rare tumors like DIPG, which lack effective treatments, the company is exploring the synergistic effects of local radiation and immunomodulation by optimizing the intraventricular administration route.

Based on research progress published in the Journal of Nuclear Medicine in September 2024, Neeta Pandit-Taskar's team innovatively evaluated the feasibility of treating DIPG with the B7H3-targeting radioimmunotheranostic agent ¹²⁴I-omburtamab via Convection-Enhanced Delivery (CED) in a first-in-human clinical study. The study enrolled 45 DIPG patients, accurately infusing ¹²⁴I-omburtamab (radioactivity 9.0–370.7 MBq) into the brain lesion area via stereotactic catheters. Continuous brain and whole-body PET/CT imaging was performed five times from 4 hours to 10 days post-infusion, alongside systematic blood sample collection, to construct a complete pharmacokinetic-dosimetric model.
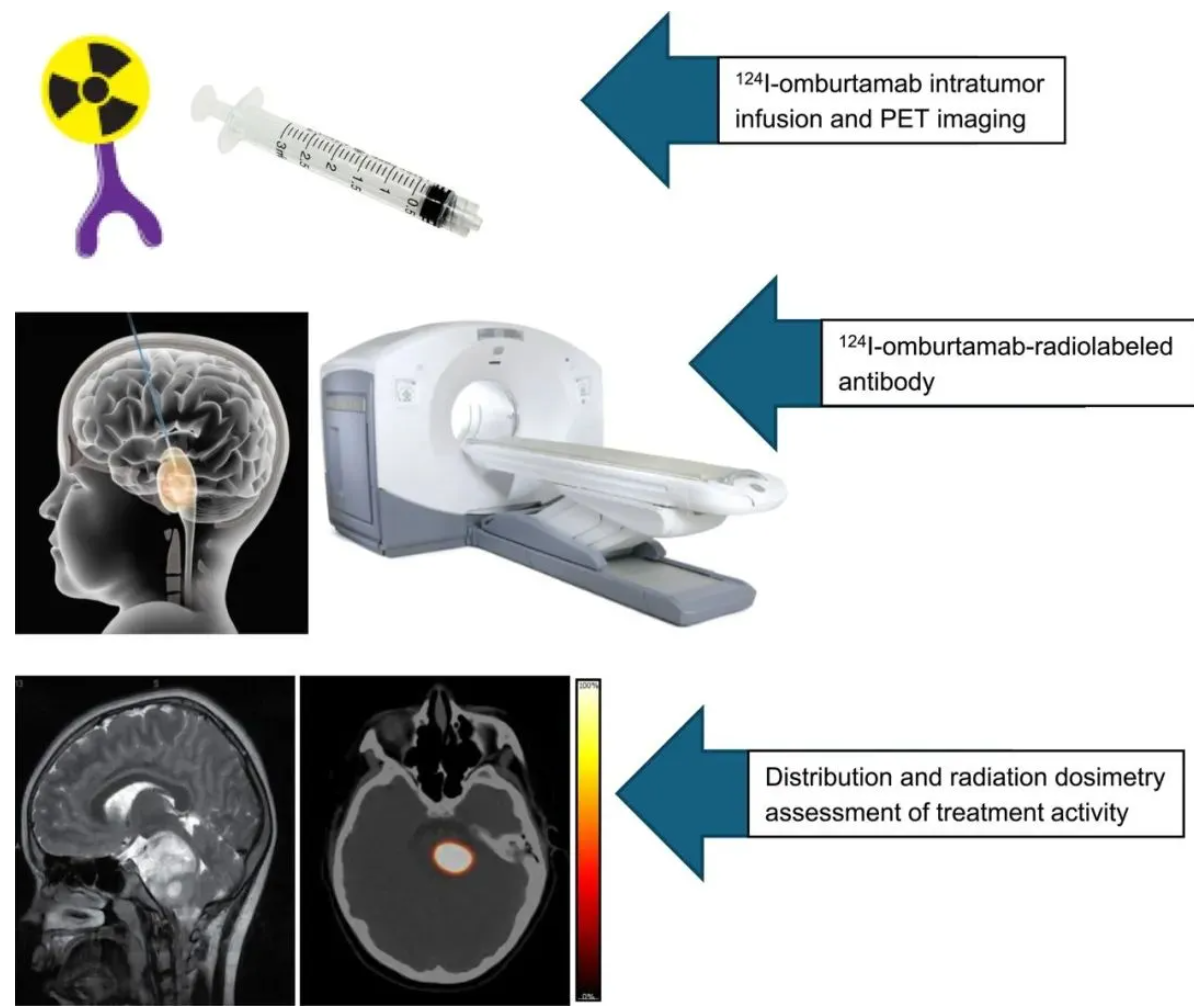
Data Source: DOI: https://doi.org/10.2967/jnumed.123.266365
Key results showed: Significant retention of radioactive activity within the tumors of all patients (mean residence time 24.9 hours, radiation dose equivalent 353±181 mSv/MBq), persisting until the last imaging (up to 10 days); extremely low whole-body and blood exposure (whole-body dose equivalent 0.69±0.28 mSv/MBq, blood dose 0.27±0.21 mGy/MBq), with minimal distribution in normal tissues; whole-body clearance followed a mono-exponential model (biological half-life 62.7 hours), while blood clearance followed a bi-exponential model ( αα -phase half-life 22.2 hours, ββ -phase 155 hours). This method achieves high-dose local tumor radiation via CED technology (significantly improving the tumor-to-normal tissue dose ratio), while real-time PET/CT monitoring confirms its theranostic potential, providing a highly efficient and low-toxicity new treatment strategy for DIPG.
 Based on a breakthrough study published in Neuro-Oncology in February 2025, Mark M. Souweidane's team pioneered the integration of a Magnetic Resonance-guided CED (MR-guided CED) system with a radioimmunotheranostic strategy, validating a precise delivery system targeting ¹²⁴I-omburtamab in 50 DIPG pediatric patients for the first time. This single-center, open-label Phase I dose-escalation trial (NCT01502917) used a 3+3 staircase design, achieving high-precision targeting of brainstem lesions through the ClearPoint® neuro-navigation system combined with Brainlab AG flexible catheters.
Based on a breakthrough study published in Neuro-Oncology in February 2025, Mark M. Souweidane's team pioneered the integration of a Magnetic Resonance-guided CED (MR-guided CED) system with a radioimmunotheranostic strategy, validating a precise delivery system targeting ¹²⁴I-omburtamab in 50 DIPG pediatric patients for the first time. This single-center, open-label Phase I dose-escalation trial (NCT01502917) used a 3+3 staircase design, achieving high-precision targeting of brainstem lesions through the ClearPoint® neuro-navigation system combined with Brainlab AG flexible catheters.
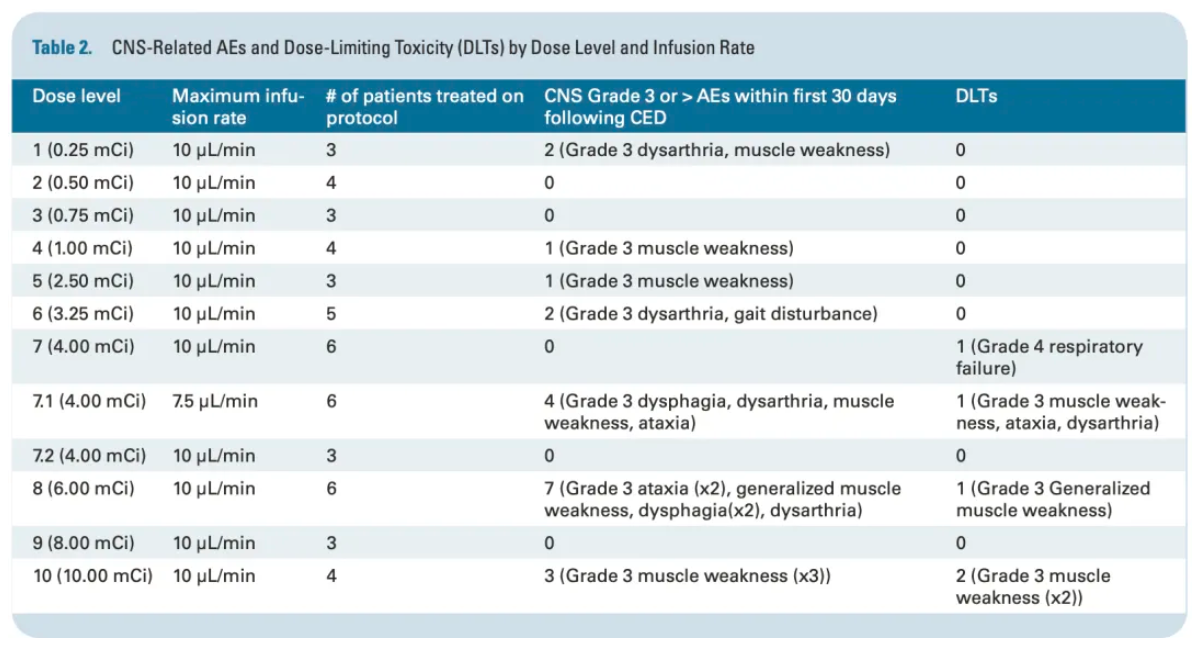 Key results showed: The maximum tolerated activity was 6 mCi (222 MBq), with an average absorbed dose to the lesion reaching 35.2±18 cGy/MBq, and a remarkably high lesion-to-whole-body radiation dose ratio of 816, indicating the characteristics of high local dose and extremely low systemic exposure. Eleven patients experienced Grade 3 treatment-related CNS toxicities (such as focal seizures, cerebral edema), and all 5 dose-limiting toxicities (DLTs) occurred in the 6 mCi dose group; no Grade 4 or 5 CNS toxicity events were observed. The median overall survival from diagnosis was 15.29 months (95% CI: 12.20-16.83), with 1-year, 2-year, and 3-year survival rates of 65.4%, 18.4%, and 11.7%, respectively, with 3 patients surviving beyond 2 years. This data validates for the first time the feasibility of ¹²⁴I-omburtamab under CED technology in improving DIPG prognosis, providing an important basis for the subsequent development of local administration strategies.
Key results showed: The maximum tolerated activity was 6 mCi (222 MBq), with an average absorbed dose to the lesion reaching 35.2±18 cGy/MBq, and a remarkably high lesion-to-whole-body radiation dose ratio of 816, indicating the characteristics of high local dose and extremely low systemic exposure. Eleven patients experienced Grade 3 treatment-related CNS toxicities (such as focal seizures, cerebral edema), and all 5 dose-limiting toxicities (DLTs) occurred in the 6 mCi dose group; no Grade 4 or 5 CNS toxicity events were observed. The median overall survival from diagnosis was 15.29 months (95% CI: 12.20-16.83), with 1-year, 2-year, and 3-year survival rates of 65.4%, 18.4%, and 11.7%, respectively, with 3 patients surviving beyond 2 years. This data validates for the first time the feasibility of ¹²⁴I-omburtamab under CED technology in improving DIPG prognosis, providing an important basis for the subsequent development of local administration strategies.

In 2025, the company announced a major strategic adjustment, establishing two business units: Radiopharmaceuticals and Danyelza. The Radiopharmaceuticals unit focuses on building the "SADA PRIT" platform, dedicated to enhancing therapeutic effects through innovative technology; the Danyelza unit concentrates on tapping into the market potential of this FDA-approved drug, which has been commercialized since 2021. Following the setback with Omburtamab, Y-mAbs quickly stabilized itself by settling with investors, restructuring its business architecture, and focusing on Danyelza, striving to stabilize its financial situation. Although these efforts briefly improved financial performance, the company's stock price failed to regain its 2020 highs, and the market remains cautious about its long-term growth potential. Whether Y-mAbs can leverage the market expansion of Danyelza and the in-depth advancement of the SADA platform to powerfully demonstrate its sustained growth potential will be the key to regaining investor confidence and opening a brand-new chapter of development.
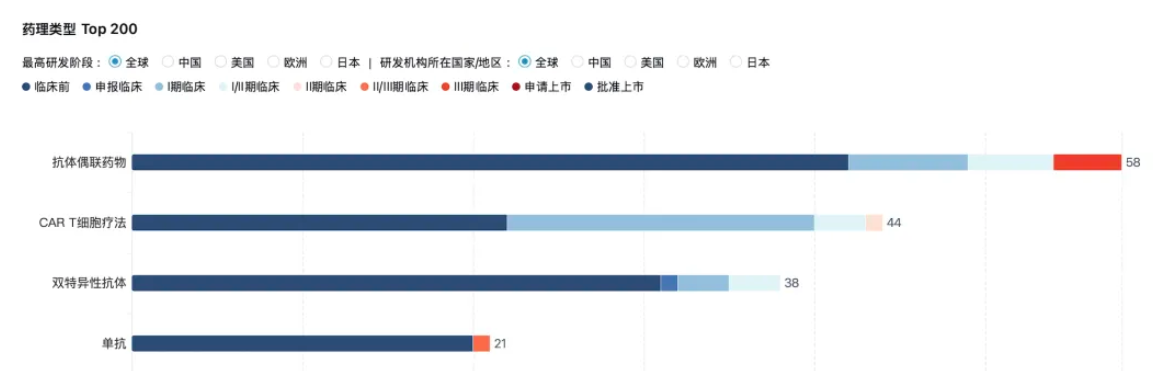
Furthermore, the scientific value and commercial potential of B7-H3 have not been tarnished by Y-mAbs. On the contrary, this target is reshaping industry cognition through breakthroughs across multiple technological pathways. Currently, continuous positive news is emerging from various development paths including bispecific antibodies, CAR-T, and ADCs. B7H3 has advanced from a "promising rising star" to a "clinical mainstay" as a target with both broad-spectrum expression and clear mechanistic potential.
 Summary: The case of Y-mAbs serves as a stark warning to those who follow: behind the "shortcut" of accelerated approval lies an even higher demand for data transparency and scientific rigor. When companies attempt to bypass the high costs and lengthy timelines of traditional randomized controlled trials by relying on single-arm studies, they must be prepared to face intense regulatory scrutiny regarding the attribution of efficacy.
Summary: The case of Y-mAbs serves as a stark warning to those who follow: behind the "shortcut" of accelerated approval lies an even higher demand for data transparency and scientific rigor. When companies attempt to bypass the high costs and lengthy timelines of traditional randomized controlled trials by relying on single-arm studies, they must be prepared to face intense regulatory scrutiny regarding the attribution of efficacy.
Address:Ping An Fortune Center, Lize Financial Business District of Beijing







